1. Introduction
Pharmaceuticals have received great consideration as aquatic pollutants due to the existence of a large number of anti-inflammatories, antibiotics, analgesics, beta-blockers, lipid regulators and anti-psychotics at concentration levels of ng L−1 to mg L−1 in municipal wastewater effluents, surface water, groundwater, sea water and even in drinking water [1–4]. The existence of pharmaceuticals in those water matrices causes aquatic toxicity due to their non-biodegradable structure. Besides, it is not possible to remove these kinds of chemicals by conventional treatment methods [5, 6]. Bearing in mind that thousands of tons of pharmaceutical constituents are manufactured and used annual in human and veterinary medicine, extensive amounts of these constituents can reach the aquatic environment, mainly by receiving wastewater from treatment plants because of their inadequate removal.
Paracetamol (PCT) (N-(4-hydroxyphenyl)acetamide) is a commonly used analgesic drug which has been found at concentrations of 7.4–45.6 μg L−1 in wastewater treatment plant (WWTP) influent in Czech Republic [3], 0.11–11.31 μg L−1 in WWTP effluent, 10.6–72.3 ng L−1 in surface water and up to 210.1 ng L−1 in drinking water in France [7].
Accordingly, there is increasing interest to develop efficient treatment processes for controlling the occurrence of pharmaceutical pollutants in aquatic bodies. Advanced oxidation processes (AOPs) have been used widely to remove pharmaceutical compounds such as ozonation [8–11], ferrate (VI) [12, 13], photocatalysis [14–16] and electrochemical oxidation [17, 18]. Chemical oxidation with ozone is an efficient technology for the removal of organic pollutants, but the performance is often not strong enough due to its selectivity to remove some target pollutants in a realistic time and cost frame. Electro-oxidation (EOX), in some cases, is an effective treatment for the degradation of biorefractory pollutants. During EOX, pollutants can be degraded by the electron transfer at the anode surface or the reaction with OH• formed from oxidation of water in acid and neutral media or OH− at pH ⩾ 10.
Another AOP for wastewater treatment is commonly used Fenton process. The oxidative potential of H2O2 can be improved in acidic medium by adding of Fe2+catalyst. According to the classical Fenton’s reaction, OH• are produced in the solution via the reaction between H2O2 and Fe2+ [19]. In recent years, instead of adding H2O2 externally, Fenton reaction has been conducted by the electrochemical approach [20, 21]. The use of carbon-based cathodes with high H2O2 production potential has attracted great attention in electro-Fenton (EF) process [22, 23].
The reaction of H2O2 with O3, i.e. peroxone process, is one of the AOPs that delivers OH• formation in aqueous solution. High reactivity of OH• allows effective pollutant degradation. As an innovative process, peroxone reaction can be conducted by the electrochemical approach and that is called electro-peroxone (E-peroxone). The E-peroxone process is a simple combination of ozonation and electro-oxidation. In the E-peroxone process, while a small amount of feeding O2 is transformed to O3 by ozone generator, the inconvertible amount of O2 is sparged into the reactor directly. Carbon–based cathode is used to convert the excessive O2 to H2O2 electrochemically. The electro-generated H2O2 then reacts with the sparged O3 to form OH•, which can remove refractory pollutants in wastewater efficiently [24]. However, in some cases, molecular O3 can be more effective species instead of OH• on the degradation of pollutants. The mechanism needs to be investigated in detail.
This work deals with a detailed study of the degradation of PCT under E-peroxone, ozonation, EF and EOX processes. As far as we know, this work reports for the first time the comparison of four different AOPs on the degradation efficiency of PCT. In this study, the effect of some experimental parameters for an individual AOPs were investigated. After the determination of the optimum conditions for each process separately, the kinetics of four different AOPs were compared for the degradation of PCT.
2. Material and Methods
2.1. Chemicals
Paracetamol Reference Standard was supplied from European Pharmacopoeia and sodium sulphate (≥ 99.99%, trace metals basis) was purchased from Sigma-Aldrich. The pH was adjusted by 0.1 M H2SO4 and 0.1 M NaOH solutions. Stock solutions for the treatment procedures (100 mg L−1 PCT) were prepared daily in high-quality pure water using the Millipore Water Purification System and stored at 4°C.
2.2. Experimental Set-up for E-peroxone, Ozonation, Electro-Fenton and Electro-oxidation Treatment of PCT
All experiments were conducted in an undivided cell with a volume of 2 L. The anode was Pt, and the cathode was carbon-polytetrafluoroethylene (PTFE) with the geometric areas of 15 and 25 cm2, respectively. Electrodes were located in the cell with a 0.2 cm gap, and initial concentration of 100 mg L−1 PCT including Na2SO4 as supporting electrolyte solution was added into the cell. The solution was stirred with a magnetic stirrer at 400 rpm during the reaction time. The current was kept constant at selected levels in E-peroxone, GEF and EOX processes. During each run, samples were taken from the supernatant layer at different time intervals and then rapidly filtered for further analysis.
During ozonation treatment, O3 from pure O2 gas (99.9%) was produced via an ozone generator (YEOJEN, OTRIO MT-20). The effluent O3 concentration in the ozone generator was adjusted by changing the ozone flow rate. The ozone generator effluent was then sparged into the cell at constant flow rates of 0.5 and 5 L min−1.
EOX and E-peroxone treatment were carried out under galvanostatic conditions using GW Instek PSP-405 Programmable DC power supply. The EOX was in the progress by turning on the DC power supply while the ozone generator was off. DC power supply and the ozone generator were both turned on simultaneously for E-peroxone treatment. GEF process was conducted at the same conditions with EOX with the addition of appropriate amount of goethite powder as an iron source to PCT solution.
2.3. Instruments and Analytical Methods
The PCT concentrations were determined by high-performance liquid chromatography (HPLC, Shimadzu) equipped with an Inertsil ODS-3 C18 column (4.6 mm × 250 mm) and a UV detector where the wavelength set at 254 nm. Acetonitrile/water (v/v) at 58/42, with the addition of 2 mL concentrated H3PO4 per liter of solution was used as the mobile phase for PCT analysis and the injection volume was 25 μL. The flow rate was 0.3 mL min−1.
Electro-generated H2O2 and aqueous O3 were monitored using a probe (JUMO GmbH & Co., Germany) during each process. The OH• concentration was analyzed as stated by terephthalic acid (TA) cumulative protocol as described elsewhere [25].
The Fe2+ and Fe3+ contents from dissolved goethite were measured using Dr. Lange cuvette tests in combination with a spectrophotometer (Hach DR-5000).
Regression coefficients (R2) and k values were determined using OriginPro 8 software.
3. Results and Discussion
3.1. The Degradation of Paracetamol by Electro-oxidation
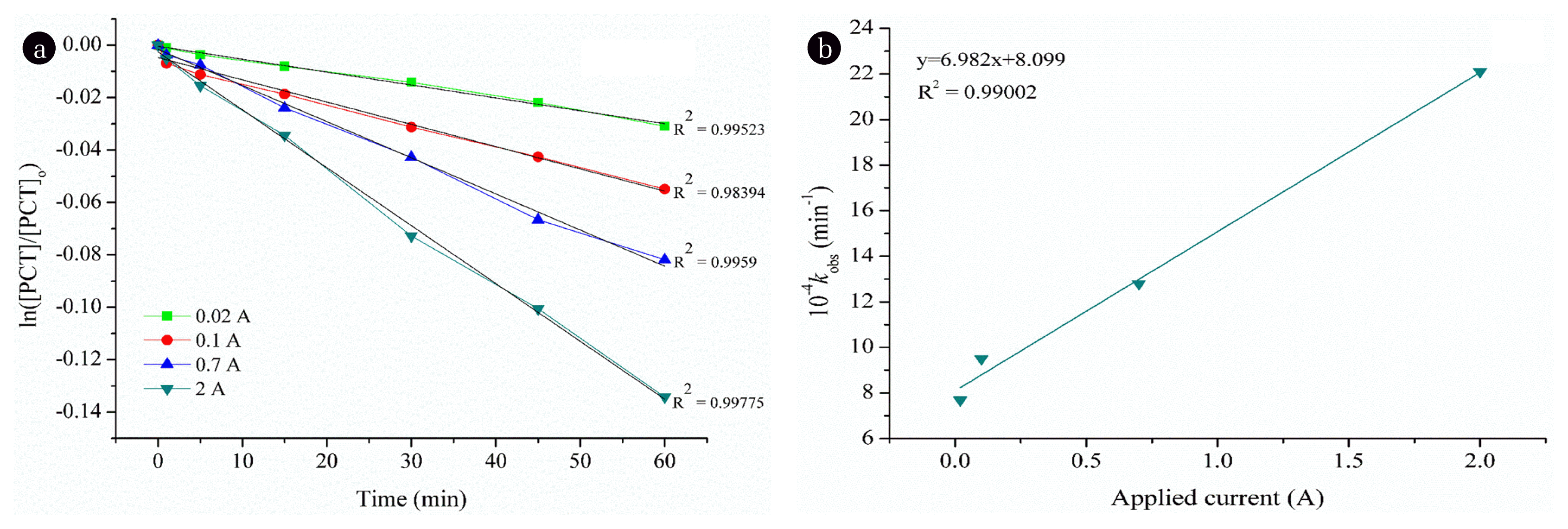
The degradation of the PCT solution by EOX at an initial pH of 6.8±0.2 was evaluated at different applied current values: 0.02, 0.1, 0.7 and 2 A. According to the results, increasing applied current enhances the degradation of PCT. However, the degradation rate was very low, even at the highest applied current. The residual concentration of PCT was 89.4 mg L−1 with only 12.9% removal efficiency.
The reaction kinetics have been studied and according to the R2 values (data not shown), the degradation of PCT by EOX process was fitted to the pseudo-first order rate law. The rate expression for the reaction of PCT with OH• may be expressed with the following equation:
where [PCT] and [OH•] are the PCT and OH• concentrations, m and n are the orders of the reaction with respect to the concentrations of PCT and OH•, respectively, and kapp is the apparent rate constant. The kinetic studies were investigated under pseudo-first order conditions with different applied current values. Eq. (1) can be rewritten as:
where kobs = kapp[OH•]n, kobs (s−1) is the pseudo-first-order kinetic constant. The degradation of PCT followed the pseudo-first-order kinetics within the studied time scales at a given applied current value as demonstrated in Fig. 1a. For a constant initial concentration of PCT (100 mg L−1), different values of kobs were determined from the slopes of linear plots at various applied currents (Fig. 1(a)). The value of kobs increased linearly (R2 = 0.9998) as applied current values were increased from 0.02 to 2 A (Fig. 1(b)), demonstrating a first-order dependence of the reaction on OH•. Accordingly, the reaction between PCT and OH• was the second order in total and first order regarding each reactant. Consequently, the rate expression for the degradation of PCT by EOX could be described as:
3.2. The Degradation of Paracetamol by Goethite Catalyzed Electro-Fenton (GEF)
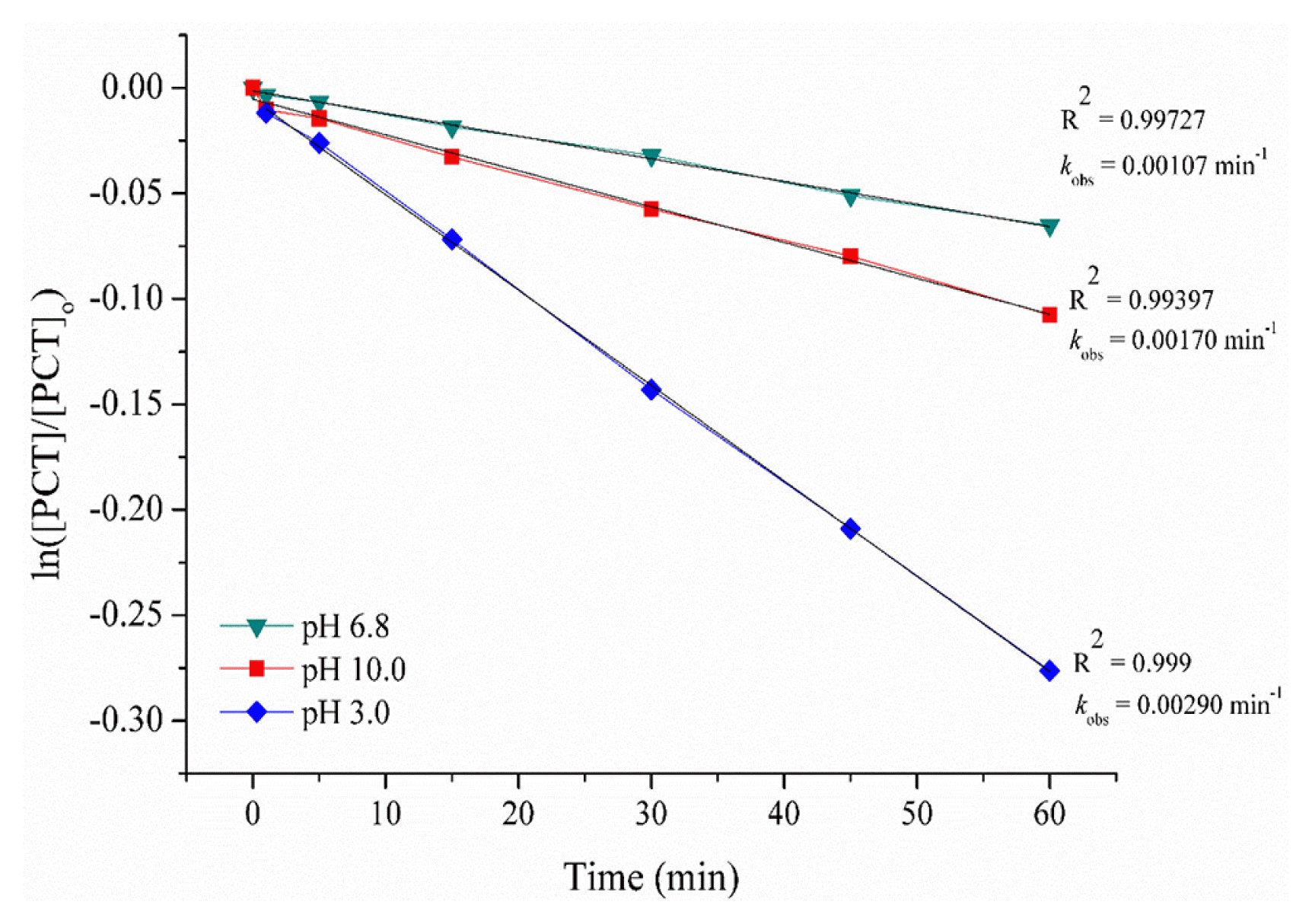
The degradation of the PCT by GEF process has been conducted at constant applied current of 0.3 A. As an iron source, 0.5 g L−1 goethite was added for each experiment. Three different initial pH values (3.0, 6.8 and 10.0) have been studied in order to understand if alkaline pH values provide better conditions or not for PCT degradation by GEF process. The results were given in Fig. 2.
As is seen in Fig. 2, acidic pH value provided more efficient and faster degradation for PCT. The concentration decays for PCT were analyzed according to the kinetic models related to different reaction orders. The best linear plot with regression coefficients of 0.999, 0.997 and 0.994 at pH values of 3.0, 6.8 and 10.0, respectively, were only found when they were fitted to a pseudo first-order model. In literature, Fenton oxidation has been usually modeled using pseudo-first order and pseudo second order kinetics in some cases. Pseudo first-order rate constants (kobs) of 0.00290, 0.00107 and 0.00170 min−1 were gained for PCT degradation by the GEF process at pH 3, 6.8 and 10, respectively. A study indicates that PCT degradation by EF process followed pseudo-first order kinetic model with rate constant of 0.6718 min−1 [26]. Even though this value was much higher than we gain in our study, the authors used only 5 mg L−1 PCT concentration. Another study indicated that pseudo-first order rate constant of PCT degradation was found as 0.0624 min−1 for UV assisted EF process [27]. t is known that acidic pH values provide more effective results in GEF process as higher pH values increase Fe(OH)3 formation and precipitation. This may cause fouling of electrode surface and prevent Fe2+ regeneration [28]. Accordingly, optimum pH value was found to be 3.0 for GEF process which agreed with previous studies [28, 29].
GEF reaction involves three key reactions as (1) the formation of H2O2 from oxygen on the cathode surface (Eq. (4)), (2) the generation of OH• via the reaction between H2O2 and Fe2+ (Eq. (5)), and (3) the degradation of target pollutant by the OH• (Eq. (6)). This is confirmed by other researchers [30, 31].
PCT can be degraded by formed OH• in electroFenton reaction, thus the reaction rate can be expressed by Eq. (7);
Here, k3[OH•] represents kobs values. Since OH• production rate is a key factor for PCT degradation by GEF process, the reaction rate can also be written considering OH• production and consumption rate. The pseudo first-order based on Eq. 7 can be defined for this process as the basic approach. From another point of view, OH• may react with Fe2+ (k4), H2O2 (k5) and Fe3+ (k6) as well [32] in addition to the above reactions (k1, k2 and k3). Also, the produced H2O2 may react with the target pollutants (k7). However, electro-generated H2O2 mainly reacts with Fe2+ to form OH• and the concentration of Fe3+ can be negligible compared to Fe2+ (see Fig. SM-1). This means k6 and k7 may not be considered. In this case, it may be assumed that OH• production and consumption rate can be described as:
In Eq. (8)k5 can be negligible to simplify the equation, because k5 was smaller than k4 according to the previous study [33]. Besides, the concentration of OH• is constant at a low level and d[OH•]/dt will approach zero concerning steady state approximation. Accordingly, Eq. (7) can also be written as:
Consequently, the rate of PCT degradation becomes a function of the PCT, H2O2 and Fe2+ concentrations.
Adsorption kinetics were also evaluated for the degradation of PCT during GEF process. The experimental data were fitted to the intraparticle diffusion model which is represented by the following Weber and Morris equation:
where, qt is the amount of absorbed pollutant (mg/g), kip (mg/g min−1/2) is the rate constant, and C is the intercept. Accordingly, the value of kip can be estimated from the slope of the plot qt versus t1/2 and qe can be calculated from the equation (qe (cal)). The experimental data were also fitted to other adsorption kinetic models such as pseudo-first order, pseudo-second order, Elovich kinetic model, and Bangham’s pore diffusion model, but the correlation coefficients obtained (data not shown) were very unfavorable. In contrast, as seen by the linear plots of the intraparticle diffusion model for all pH values given in supplementary material (Fig. SM-2), there is a good fitting of the experimental results to the model. Indeed, as indicated in Table 1, the correlation coefficients are higher than 0.99 and the experimental and calculated qe values (qe (exp) and qe (cal), respectively) are very similar.
It can be said that PCT adsorption onto goethite was diffusion controlled since intraparticle diffusion model agrees well with the experimental data (the plot of qt against t1/2 was linear for all pH values) and intraparticle diffusion was the only rate-limiting step. Also, the adsorption process was faster in pH 3 compared to pH 6.8 and 10 as expected.
3.2. The Degradation of Paracetamol by Ozonation
OH• can be generated through the reaction between O3 and OH− during ozonation. The generation of H2O2 in the form of its conjugated base, HO2−, is also observed during ozonation process (Eq. (11)). Produced HO2− may then react with O3 to form OH• (Eq. (12)).
Therefore, in this case, PCT can be degraded either directly by O3 or indirectly by OH•. Consequently, the kinetics of PCT degradation by ozonation can be expressed as follows:
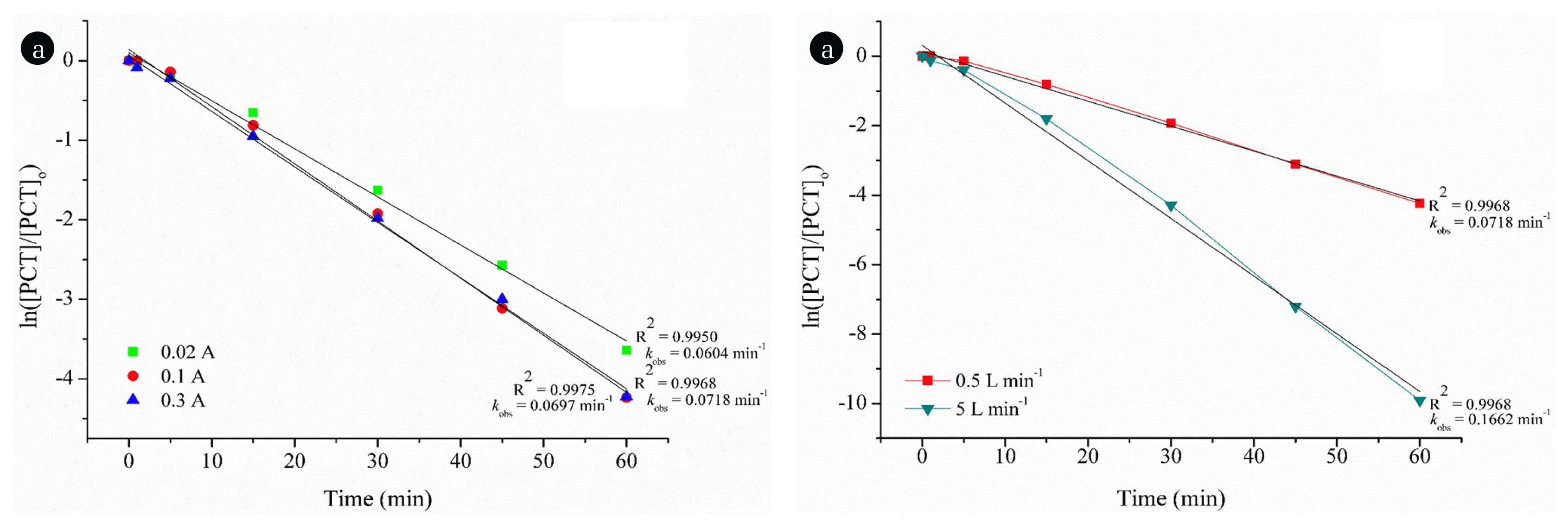
The reaction kinetics has been calculated along with the R2 values (data not shown) and the degradation of PCT by ozonation process gave the highest R2 value when it was fitted only to the pseudo-first order rate law. The observed rate constants (kobs) were given in Fig. 3(a) according to ozone flow rate.
O3 and OH• concentrations besides the corresponding rate constants are needed to predict PCT degradation by ozonation. O3 concentrations can be easily determined with standard methods. However, OH• concentrations are difficult to measure due to their very short half-life in waters. Then, the Rct model in which the Rct is determined by the ratio of OH• exposure to O3 exposure (Rct = ) can be used [34]. As stated by Elovitz and von Gunten [34], Rct is constant in water for the duration of ozonation after early phases. Hence, Rct can be written as the ratio of the [OH•] to the [O3]. So, Rct × O3 is written instead of [OH•] in Eq. (13), then it yields Eq. (14):
The pseudo-first-order kinetic constant can be written as;
In Fig. 3(b), the effect of O3 exposure on the degradation of PCT was also given. As seen, almost complete degradation of PCT was observed at the end of reaction time with the application of 5 L min−1 ozone flow rate.
3.2. The Degradation of Paracetamol by E-peroxone
The degradation of PCT by E-peroxone process has been studied at an initial pH of 6.8 and the effect of applied current on the E-peroxone treatment of PCT has been assessed (Fig 4(a)). As illustrated in Fig. 4(a), increasing applied current to 0.1 A from 0.02 A enhances the degradation of PCT. However, further increase in current almost did not change the degradation rate.
The increase in applied current can provide more aqueous OH• production if sufficient aqueous O3 exists in the solution. The key point is that to define the critical applied current value due to low solubility of O3. In other words, OH• formation would be limited by means of O3 transfer rate from gas to liquid phase as the current increases the critical value. Besides, H2O2 is known to be OH• scavenger. Thus, increasing applied current beyond a critical value may produce more H2O2 than needed, which can diminish the E-peroxone process efficiency [35, 36]. Consequently, the critical applied value was determined as 0.1 A in this case.
The effect of O3 flow rate has been investigated for E-peroxone degradation of PCT. As seen in Fig. 4(b), increasing O3 flow rate considerably increased the degradation rate of PCT. Complete removal was observed with O3 flow rate of 5 L min−1 at the applied current of 0.1 A.
OH• is generated through the reaction between O3 and H2O2 during the E-peroxone process [37–39]. Therefore, the degradation of PCT depends on the concentrations of O3, H2O2 and OH•. However, O3 reacts with H2O2 only in the form of its conjugated base, HO2−, and then the generation of OH• takes place:
The reaction kinetics have been considered by the highest R2 values (data not shown) for the degradation of PCT by E-peroxone process and the highest R2 value was obtained when it was fitted to the pseudo-first order rate law. kobs values were determined as 0.0718 and 0.1662 min−1 for O3 flow rates of 0.5 and 5 L min−1, respectively at the applied current of 0.1 A. In E-peroxone process, PCT is mostly degraded by OH•:
Here, kOH[OH•] indicates kobs values. From a deeper point of view, O3 may also contribute the degradation of PCT, however, O3 reacts primarily with electro-generated H2O2 to produced OH•. In this case, the kinetics of PCT degradation by E-peroxone can also be expressed as follows:
Some amount of O3 is consumed according to the Eq. (17). Hence, O3 consumption also should be considered;
3.5. The Comparison of the Processes on the Degradation of PCT and Formation of Intermediates
The degradation of commonly used analgesic PCT was evaluated by different AOPs in this study. In overall assessment, EOX, GEF, ozonation and E-peroxone processes were compared in terms of PCT degradation. All studied processes showed pseudo-first order reaction kinetic for PCT degradation. The conditions that gave maximum PCT removal for each process were determined and kobs values for those circumstances of EOX, GEF, ozonation and E-peroxone processes were 0.0022, 0.0029, 0.0870 and 0.1662 min−1, respectively (See Table 2). The order of the processes was E-peroxone > ozonation > GEF > EOX for PCT degradation at their individual optimum conditions.
The complete degradation of PCT was observed in E-peroxone process. Due to the high rate constants at the E-peroxone and ozonation, PCT degradation would mostly take place by OH• and molecular O3. In contrast, with lower kobs values obtained in EOX process, hydroxyl radicals would not contribute considerably to the PCT degradation. There was almost no production OH• during EOX process in the bulk solution. Direct anodic oxidation via electron transfer on the electrode surface and produced OH• on the electrode surface (Pt(OH•)) could be the main mechanism for EOX processes at Pt anode. In GEF process, relatively lower kobs values were also obtained comparing E-peroxone and ozonation processes. This was because of lower OH• production rates. However, GEF process provided slightly higher efficiency in PCT degradation than EOX process. This can be concluded that having an additional source of oxidizing reagents provides more OH• production in the bulk solution. Besides, it should be noted that the applied current was 2 A in EOX process where it was 0.3 A in GEF process. It can be said that additional iron source to EOX process helps to reduce applied current and as well as energy consumption.
In Fig. 5, H2O2 and OH• production capacities can be seen. Accordingly, H2O2 production increased during the time for GEF, ozonation and E-peroxone. Although the same electrodes (carbon-based cathode and Pt anode) were used in EOX process, negligible amount of H2O2 was produced. However, the addition of goethite in GEF process increased the amount of produced H2O2. It is known that H2O2 is produced at carbon-based cathode. However, the addition of iron source may contribute and increase the formation of H2O2 according to the following equations [19, 40]:
The capacity of H2O2 production was extremely high at the end of time in ozonation and E-peroxone compared to GEF process. At 60 min of reaction time, 468.0, 2168.3, and 2675.0 mg L−1 H2O2 was produced in GEF, ozonation and E-peroxone, respectively. However, H2O2 production rate was higher in the middle stages of GEF process compared to ozonation and E-peroxone. For instance, between 30–45 min of process time, while H2O2 concentration ranged between 200–344 mg L−1 for EF, it ranged between 161.52–323 mg L−1 and 183–190.6 mg L−1 for ozonation and E-peroxone, respectively. This agrees with the above conclusion about less efficiency of the GEF process by means of excess H2O2 behaved as scavenger of OH•. As it is seen from Fig. 5, when H2O2 production increased OH• formation decreased.
When OH• production capacity was compared, OH• formation increased until 10, 3 and 5 min of reaction time in GEF, ozonation and E-peroxone, respectively. Then, a decreasing trend was observed for all the processes. In EOX, OH• production was almost negligible comparing the other processes. Up to 0.87, 23.32, and 20.64 μM of OH• concentrations were produced in GEF, ozonation and E-peroxone, respectively. Ozonation and E-peroxone processes provided much higher OH• production rates in contrast to GEF and EOX processes. This result agreed with the above conclusions.
Energy consumption should also be considered for the comparison of the processes. For this reason, specific energy consumption (SEC) values were calculated considering 50% PCT removal percentage for each process according to the following equations:
Where, U is the average voltage (V) recorded during the processes, i is the applied current (A), t is the process time (h), [PCT]0 and [PCT]t represent the initial and treated PCT concentrations, respectively and V is the solution volume (L). r is the energy need for ozone production (15 kWh kg−1) [33], and CO3 is the amount of consumed ozone (g) which was calculated according to Eq. (24):
Where Qg is the gas flow rate (L min−1) and [O3]inlet is the inlet O3 concentration (mg L−1). The results and the conditions for each process can be found in supplementary material (Table SM-2). As seen from Table SM-2, SEC values were determined as 7.7844, 0.7158, 0.1452 and 0.1164 kWh gPCTremoved−1 for EOX, GEF, ozonation and E-peroxone, respectively. This suggests that ozonation and E-peroxone processes were cost effective compared to the other two processes. In conclusion, ozonation and E-peroxone processes were found to be more effective for PCT degradation considering process efficiency and energy consumption.
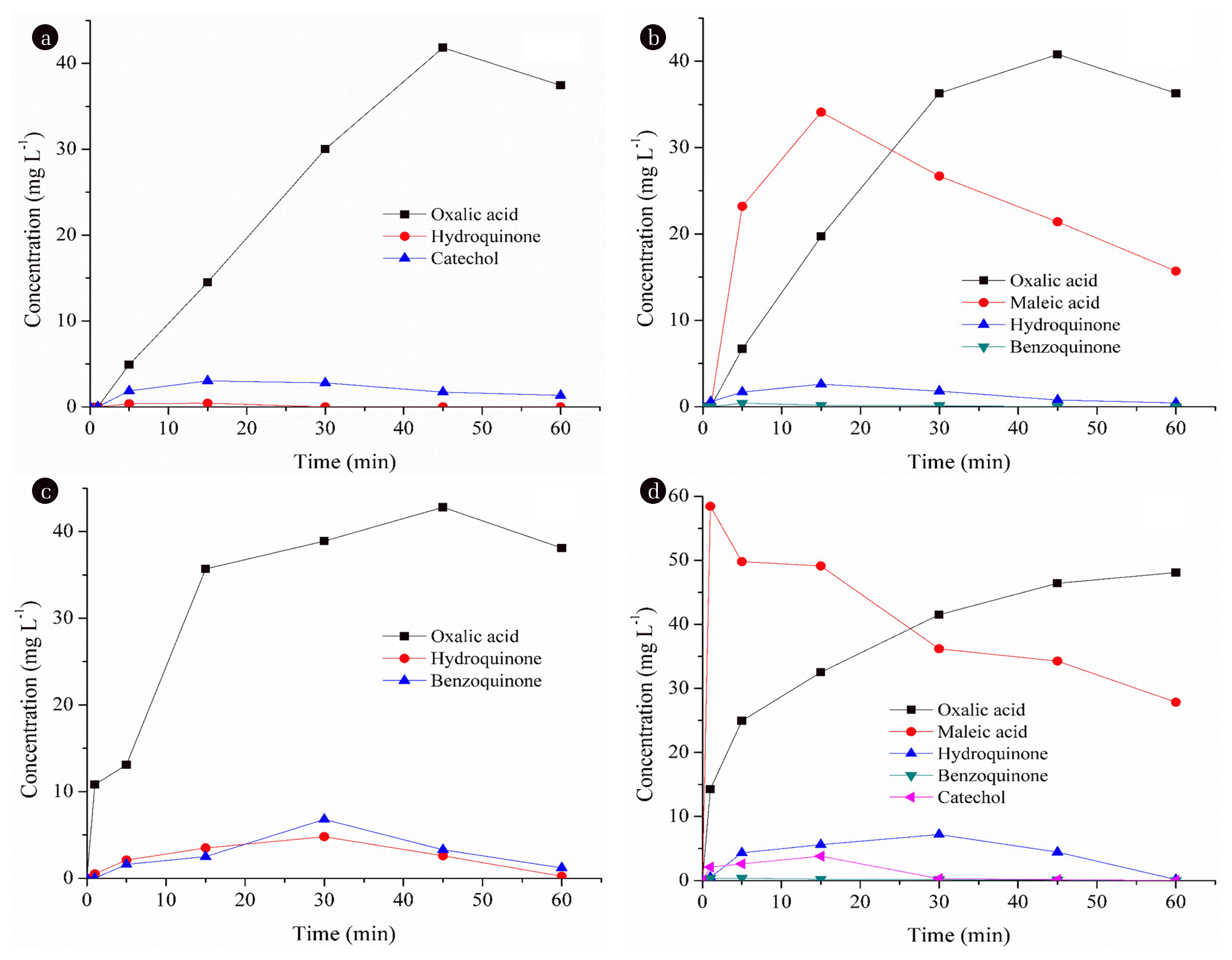
The formation of intermediates during each process is given in Fig. 6. As can be seen in Fig. 6 (a), only hydroquinone and catechol as aromatic compounds were identified during E-peroxone process indicating that hydroxyl radicals attacked in the aromatic ring of PCT. The concentration of hydroquinone was very low during the process and it almost disappeared at 30 min of process time. This suggests that the aromatics rapidly destroyed on the anode and were not accumulated in the medium. The degradation of PCT occurred via the formation of oxalic acid which come from the destruction of benzenic ring of aromatic compounds during E-peroxone process. During ozonation of PCT (see Fig. 6(b)), hydroquinone and benzoquinone were identified and they almost completely destroyed at 60 min and 15 min of process time, respectively. Final products were harmless carboxylic acids (oxalic acid and maleic acid). Oxalic acid concentration increased up to 45 min and then decreased, while maleic acid concentration increased until 15 min of process time and then decreased. During GEF process, aromatics (benzoquinone and hydroquinone) and oxalic acid formed (see Fig. 6(c)). The concentration of aromatics increased until 30 min of process time and then decreased. Aromatic compounds (benzoquinone, hydroquinone and catechol) and carboxylic acids (oxalic and maleic acids) were identified during EOX process (see Fig. 6(d)). However, the concentration of benzoquinone was very low during the EOX process and hydroquinone and catechol almost disappeared at 60 min and 30 min of process time, respectively. The final products were oxalic acid and maleic acid during the degradation of PCT by EOX process. As seen in Fig. 6, carboxylic acids (oxalic and/or maleic acid) were final products for all processes during the degradation of PCT generated by ring cleavage and decarboxylation reaction which was reported by other researchers [41–43].
4. Conclusions
The degradation of PCT by EOX, GEF, ozonation and E-peroxone has been investigated in the present study. The higher and faster PCT degradation efficiencies were observed during E-peroxone and ozonation treatment. Kinetic studies showed that all processes were kindly fitted to pseudo-first order kinetic for PCT degradation. kobs values were determined as 0.00221, 0.0017, 0.0870 and 0.1662 min−1 for EOX, GEF, ozonation and E-peroxone processes, respectively. For GEF process, the adsorption part was controlled by diffusion. Where OH• production was almost negligible in EOX, up to 0.87, 23.32, and 20.64 μM OH• was generated in GEF, ozonation and E-peroxone, respectively. Ozonation and E-peroxone processes delivered considerably higher OH• production rates, contrary to GEF and EOX processes. When kobs values of each process are compared, E-peroxone and ozonation processes which O3 is dominant, have much higher rate constants comparing EOX and GEF. Thus, it can be concluded that PCT degradation would mostly occurs through OH• and molecular O3. On the contrary, the EOX process with lower kobs values, hydroxyl radicals would not contribute noticeably to the PCT degradation. In the point of energy consumption, E-peroxone and ozonation were cost effective processes compared to EOX and GEF processes on PCT degradation. Aromatic compounds and carboxylic acids were identified as degradation intermediates during the processes and their concentration varied for each process. However, final by-products were carboxylic acids for all processes.