Physical and chemical Laboratory of materials, catalysis and environment (LPCMCE), University of sciences and technology of Oran Mohamed Boudiaf USTOMB 1505 Oran,
Algeria
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
In this work, a detailed study on the electrochemical degradation of an azo dye, Orange G is performed using a platinum electrode. Indeed, the influence of the dye concentration (50–150 mg/L), the pH of the medium and the density of the electric current is studied on the rate of discoloration, the rate of mineralization, the efficiency of the electric current and the energy consumption. The UV-visible spectra of OG plotted against the degradation time show the decrease of the intensity of the characteristic dye peaks. In an environment rich in chlorides, all peaks disappear after 15 min of degradation. However, the peaks at wavelengths of 200 and 290 nm appeared after one hour of treatment. In K2SO4, the eliminated percentages are respectively 46, 54 and 61% for wavelengths of 245, 330 and 480 nm. This suggests that the degradation mechanisms in K2SO4 and KCl environments are not the same. In the middle rich in chlorides, the eliminated percentage of OG did not seem to be affected by the concentrations increase. These results confirm the hypothesis that electrochemical oxidation process is very favorable for concentrated pollutants discharge.
Nowadays, electrochemical methods of water treatment are very important because of their high efficiency, their ease of handling and low cost [1–3]. Several studies have shown the reliability of electrochemical techniques for treating synthetic industrial discharges containing several compounds. The results of their research enabled the determination of the experimental conditions necessary to remove of organic matter from the actual effluents [4–6]. Despite the advantages outlined above, some deficiencies persist, limiting the use of these electrochemical methods in the treatment of industrial discharges. Examples of these negative points include the resistance of the anode material to degradation (corrosion), its high cost and the low efficiency of the current [7, 8]. In conclusion, an extensive research needs to be done to determine several key parameters such as: the effect of pH, the type of pollutant, its conductivity and concentration, and the design of the electrochemical reactor (anode material, electrode arrangement, appearance adverse reactions [9].
Platinum is among the most used materials in electrochemistry. Its good chemical resistance to corrosion as well as its stability even in highly aggressive media has encouraged its use in the electrochemical degradation of pollutants. The results presented by different researchers, showed a very interesting electrocatalytic activity in this field [10, 11]. As is known, Pt has strong dehalogenation ability [12], which is helpful to the degradation of halogen aromatic hydrocarbons. However, the disadvantage of Pt is its relatively low oxygen evolution potential (ca. 1.2 V). Tabbara et al. [13], have studied the discoloration of Eosin Y and Rose Bengal by indirect anodic oxidation by employing boron doped diamond (BDD) and Pt electrodes. The discoloration at BDD electrode in presence of KCl is similar to that at Pt electrode, since oxidation of chloride occurred at the 2 types of electrodes [13]. Electrochemical oxidation of reactive textile dyes using platinum anode and Na2SO4 was realized. After 60 min of electrolysis, complete degradation of dyes was reached (COD value below 30 mg/L O2) [14]. There is much work on dye degradation [15, 16]. However, there is no available report on the electrochemical degradation of OG using a platinum electrode.
In this study, the anodic oxidation of OG using the platinum anode is investigated in order to analyze the efficiency of anodic oxidation of OG under different operating conditions. The influence of pH, current density and dye concentration on discoloration, degradation rate, mineralization, current efficiency and energy consumption is examined.
2. Experimental
2.1. Reagents and Chemicals
The dye used in this study Orange G (OG) is provided by Across Organics (Table 1). All the other reagents such as anhydrous K2SO4, KCl NaOH and H2SO4 are of analytical grade with purity level > 98% from Sigma–Aldrich and Merck.
2.2. Procedures and Equipment
The electrochemical experiments were conducted at room temperature in a one-compartment electrochemical cell. To supress the influence of mass transfer rate of pollutants to the electrodes on the removal efficiency, high mixing was provided with a magnetic stirrer. The cell contained two parallel planar electrodes with an inter-electrode distance of 10 mm, using platinium electrode as anode (10 mm2) and Platinium wire as cathode (Fig. 1). The anode potential was monitored during the runs using a reference electrode (saturated Ag/AgCl) connected to the working electrode. All degradations were carried out under galvanostatic conditions by the galvanostat (Radiometer analytical PGZ 301), for a volume of 100 mL of wastewater in 0.1 M K2SO4 or KCl inside a standard three-electrode cell at room temperature. The degradation started as soon as the electricity was switched on and the current was set at the desired value. The pH variation was recorded by a pH meter (HANNA HI 8521). The samples were taken at given time intervals for analysis.
The UV-Vis spectra of OG were recorded in 200–800 nm range using the UV-Vis spectrophotometer (Specord 210 Analytik Jena). The decolorization ratio was calculated by the change of absorbance using the following formula:
(1)
Where A0 and At are the absorbance of OG at the initial and given time t, respectively.
The specific energy consumption (Ec, in kWhm−3) was calculated as follows,
(2)
Where, U is the average cell voltage (V), I the current (A), t the treatment time (s) and V the volume of the treated wastewater (L).
The Chemical Oxygen Demand (COD) was determined by oxidation with dichromate following the standard methods. Before the analysis of COD, samples were diluted (1:4) to maintain the concentration of chlorides below 2 g.L−1.
The general current efficiency (GCE) was determined using the relation:
(3)
where, COD0 and CODt are the chemical oxygen demand at initial and given time t (in g O2 L−1), respectively, F the Faraday constant, and other parameters are stated the same as above. GCE is defined as the ratio of current used in oxidizing pollutants to the total current in the circuit. The performance of all the treatment processes was followed by the analysis of colour and COD at different time intervals, up to 240 min.
3. Results and Discussion
3.1. Effect of Current Density
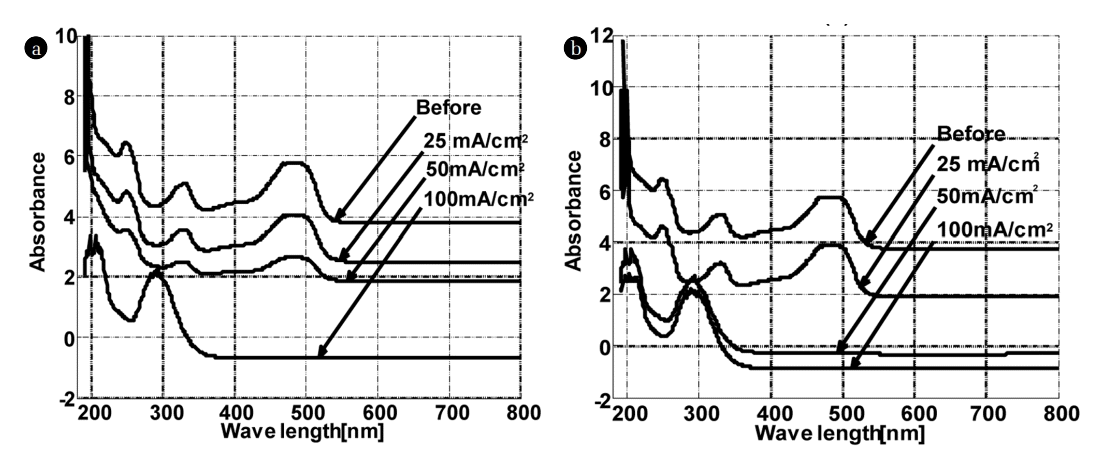
The experimental UV-visible spectrum of Orange G (50 mg.L−1) (Fig. 2) shows that it consists of 4 bands of variable intensities located at 220, 245, 330 and 475 nm, respectively. Indeed, the researchers explained the characteristic bands of orange G as follows: The band centred at 330 nm absorbing relatively at high energy, is attributed to HOMO-7 -> LUMO transition which derives from π–π* transition between π system of naphthalene ring, the SO3 groups and the π* system of the – N=N- azo link as discussed previously elsewhere [17]. In contrast, the strong band centered at 475 nm absorbing relatively at low energy, is assigned to the HOMO-1 -> LUMO (71%) and HOMO-2 -> LUMO+1 (17%) electronic transitions. The LUMO is principally the π* system of −N=Nazo link, while the HOMO and HOMO-1 are mainly localized on SO3 groups. It is noteworthy to mention that chemical structure of this dye is composed of a conjugated system allowing a more increasing intensity of these transitions, producing thus, the colour of this system. Moreover, natural band analysis indicates that molecular orbital are mainly composed of π atomic orbital, so, the electronic transitions are mainly derived from the contribution of π–π* bands [17, 18].
The current density is an important variable in electrochemical processes. The comparison between the UV - visible spectra of the dye before and after treatment shows the decrease in the absorption peaks intensities at 245, 330 and 475 nm with current density (Fig. 2).
During this time, the colour of the OG solution changed from orange to almost colourless.
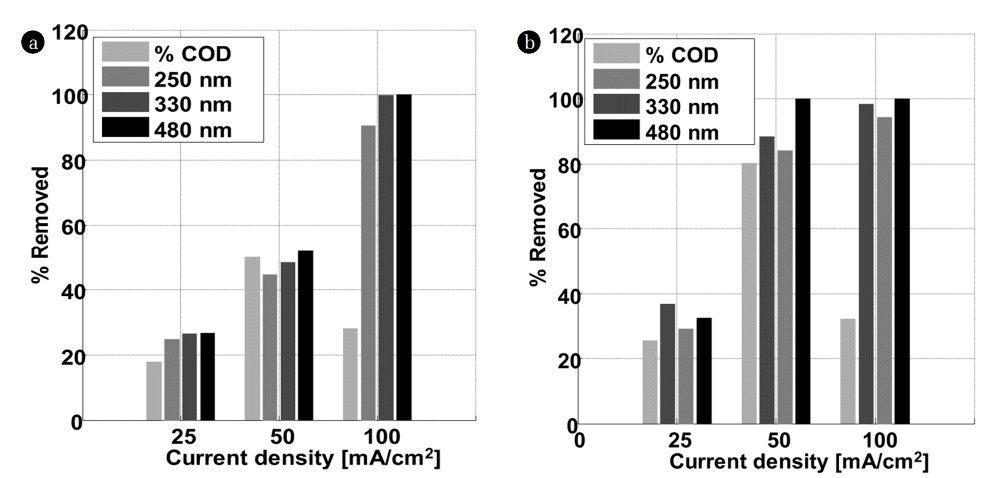
The plot of the percentage of OG removed at the characteristic wavelengths (245, 330 and 480 nm) and that of COD (Fig. 3) removed following the electric current density, confirms the results mentioned above. In K2SO4, the electric current density has no influence on the peak at 245 nm. In contrast, the best removal was obtained at 100 mA/Cm2 for the peaks located at 330 and 480 nm. In the middle rich in chlorides, the total elimination of the three peaks (245, 330 and 480 nm) starts from 50 mA/Cm2, the best results were also recorded for 100 mA/Cm2. Based on these results, one can deduce that the colour disappearance is closely related to the peaks disappearance at 330 and 475 nm. These results are in agreement with those of Yoshida and co-workers relative to the discoloration of an azo dye on a palladium electrode [19]. The colour of OG originates from double bonds associated with an azo group (N = N) [20]. The peak around 245 nm is attributed to the aromatic rings and the unsaturated groups in the molecule of the dye. All these findings suggest that the unsaturated bonds are conjugated and broken; the molecule of Orange G may be divided into small molecules after electrochemical oxidation. Besides, the appearance of a new peak at 290 nm after electrochemical degradation was noticed for 100 mA/Cm2 and 50 mA/Cm2 in K2SO4 and KCl, respectively.
These results are confirmed by measurement of COD removed depending on the intensity of the current used (Fig. 3). By increasing the electric current density from 25 to 100 mA/cm2, the percentage of COD removed in K2SO4 and KCl increases from 17 to 28% and from 25 to 32%, respectively. The best results were obtained for 50 mA/cm2 of current density. The percentages of COD eliminated, are 50 and 80% in K2SO4 and KCl, respectively. These results are in agreement with the literature; some researchers in the field found that the percentage of COD eliminated, increases in the presence of chloride ions [21]. Panizza found that the electric current density increases until it reaches a certain value, called the limiting current density. The percentage of removed COD does not increase because of the mass transfer limitation [22]. Beyond this value, the electricity supplied to the system does no longer serve to oxidize the dye. Nevertheless, it produces oxygen gas. On the other hand, the formation of polymeric products is favored by high current densities making the removal of COD difficult.
3.2. Effect of pH on the Degradation of Orange G
The pH is an important factor for release treatments. In the anodic oxidation, there are several works on the influence of pH. Unfortunately, results differ from a search to another because of the difference in the working conditions (electrode material and pollutant) [23]. Fig. S1 shows the variation of the percentage of OG removed at 480 nm with the pH in absence and presence of chloride ions. For a dye concentration of 50 mg/L, a current density of 50 mA/cm2 and a treatment time of 4 h, the percentage of dye removed is almost identical regardless the initial pH of the solution. Similarly, the potential of the electrochemical cell does not change with pH. It is known in electrochemistry that the redox potential of the majority of organic products is affected by the change solution pH. In most cases, the efficiency of oxidation is higher in acidic media. This is not the case in this study. The reason may be that OG is a kind of conjugated azo compounds containing benzene ring. This may be the same reason which leads to little dependence between electrochemical oxidation of OG and the pH value of the medium. Wu and al. [21] for degradation of MB onto BDD in chloride mediated and chloride free water observed similar trend. Zhou et al. [24] compared the electrochemical degradation of a model azo dye of methyl orange on the mixed metal oxide (MMO) and boron-doped diamond (BDD) electrode in Na2SO4 and NaCl. They confirmed that the effect of pH on the decolorization on BDD was not significant, which was in agreement with others’ observation on orange II oxidation on BDD electrode [24]. This indicates that the degradation of Orange G on a platinum electrode is feasible in a wide pH range. It would be, therefore, useless to adjust it for a possible application of the method in the industry.
3.3. Degradation Kinetics of Orange G
The degradation kinetics of Orange G on a platinum electrode is followed by the analysis of UV-visible and COD in two different media: Firstly in non-chloride medium (K2SO4) and secondly in the presence of chloride ions (KCl) for OG concentration of 50 mg/L, a current density of 50 mA/cm2 and 4 h of treatment time.
The UV-visible spectra of OG plotted against the degradation time show the decrease of the intensity of the characteristic peaks of dye with time. In the middle rich in chlorides, all peaks disappear after 15 min of degradation. In addition to this, we note the appearance of two new peaks at 200 and 290 nm after one hour of treatment (Fig. S2). This suggests that there is a possibility of formation of other products. The dye removal percentages at characteristics wavelengths (245, 330 and 480 nm) are 100, 90 and 83%, respectively
In K2SO4, the eliminated percentage increases with the increase of the treatment time for the three characteristic wavelengths of the dye without reaching equilibrium. After 4 h of treatment, the percentages eliminated are, respectively 46, 54 and 61% for wavelengths of 245, 330 and 480 nm (Fig. 4).
The results presented previously are confirmed by measurement of COD eliminated percentage according to time (Fig. 5). In effect, after 2 h of treatment, 31% in K2SO4 and 68% in KCl are eliminated. These results are in agreement with those of Panizza et al. [25], who found that the electrochemical oxidation of the methyl blue by Ti / TiRuO2 electrode in the presence of NaCl electrolyte allowed fast mineralization compared to that made in absence of chlorides ions [25]. After 4 h of treatment, the percentage of eliminated COD reaches 50% in K2SO4 and 80% in KCl. This can be explained by the accumulation of carboxylic acids as final products [26].
The electrochemical degradation mechanism on a platinum electrode is essentially due to:
1. Free radicals produced by oxidation of water (direct oxidation) [9]:
(4)
A high decolorization efficiency, but with very low decontamination, is a result of the nature of Pt electrode, which is an active anode that accumulates small quantity of physisorbed Pt(OH•) on its surface during water discharge. This behaviour is found for most dyes given in the literature [27], indicating that Pt anodes are efficient electrocatalytic materials when it comes to removing color, but without a satisfactory organic matter removal. However, the poor oxidation action of Pt(OH•) in sulphate or acidic media can be enhanced, in some cases, by using NaCl as supporting electrolytes or by adding a known amount of NaCl to the solution [28]. This gave much better performance due to the additional oxidation of organics with active chlorine species [29].
2. Indirect oxidation in the presence of chloride ions made by the chlorine active species (HClO and Cl2) generated by the following reactions [30]:
(5)
(6)
(7)
Regarding the anode material, its non-active or active nature is an important parameter that determines the predominant category of oxidants produced during the electrolysis of chloride solutions. Non-active anodes like BDD are not useful for indirect electro oxidation with active chlorine since they form largely ROS and other oxidants like peroxodisulfate, peroxodicarbonate or peroxodiphosphate [9], avoiding a good active chorine species generation. Conversely, active anodes have much higher electrocatalytic power for oxidizing chloride ion than for generating ROS, as found for DSA-type [31], Pt [32] and graphite electrodes [33]. Once the species of active chlorine are transferred in solution, they provoke a series of reactions and accelerate the process of degradation. Thus, the electrochemical oxidation of OG is improved by both action of oxidizing specie: the free active radicals and chlorine one. Besides, because of the big concentration of the chloride ions in industrial wastewater this technique can be promising for the treatment of the colourful releases.
In the literature, the kinetics of electrochemical oxidation of most dyes follows the pseudo first order [30, 34].
(8)
Where C is the concentration of dye in mg/L and kobs the apparent rate constant of the reaction.
In this work, the kinetic’s degradation of Orange G at different concentration (50, 75 and 100 mg/L) using a current density of 50 mA/cm2 in the presence and absence of chloride ions seems to follow the pseudo first order except for the concentration of 100 mg/L in KCl. The results are shown in Fig. S3. The values obtained for the pseudo first-order kinetic constants at different concentrations are summarized in Table 2. In K2SO4 medium, the pseudo first order rate constant is the same for the three concentration of OG (50, 75 and 100 mg/L). This very low decolorization efficiency in K2SO4 is a result of the nature of Pt electrode, which is an active anode that accumulates small quantity of physisorbed Pt(OH•) on its surface during water discharge. This behaviour is found for most dyes which are given in the literature [27]. However, the poor oxidation action of Pt(OH•) in sulphate media can be enhanced, by using KCl as supporting electrolyte, which gave much better performance due to the additional oxidation of organics with active chlorine species [28, 29].
As it is explained previously, the degradation of dyes in the presence of chloride ions is made by the simultaneous action of direct and indirect oxidation. The indirect role of the oxidation by the active species of chlorine ions can be estimated using the following formula [24]:
(9)
Where kobs1 and kobs2 are the apparent rate constants for the electrochemical degradation of Orange G in KCl and K2SO4, respectively. The results are shown in Table 2.
The presence of chloride ions in solution greatly improves the rate degradation of Orange G. Comparison between kinetic constants demonstrates this behavior. It is clear that under these conditions, the role of indirect oxidation exceeds 55 times that of the direct oxidation for 50 mg/L of OG concentration. Zhou and co-workers found a factor of 73.3 for the oxidation of orange II on a BDD electrode [24]. It is observed from the results (Table 2) that the rate constants of OG degradation in KCl medium, decreased with the increase in initial OG concentration. These can be explained in terms of diffusion control, assuming that mineralization occurred on the electrode surface mediated by OCl− ions. At low initial concentrations, the electrochemical reaction is faster than the diffusion. When the initial concentration increases, more organic molecules are transferred to the surface of electrode and the amount of dye reduction is increased. The OCl- ions that are generated are limiting in this case, and the degradation efficiency would decrease with increased initial concentration of the dye [9]. Unfortunately, when the concentration of the dye reached 100 mg/L in KCl medium, the pseudo first order is inappropriate to pose the degradation results. On the other hand, the pseudo-second order kinetic model ((1/A0)-(1/At) vs. time) in the KCl show suitable linearity compared to pseudo-first order model. The coefficient of pseudo-second order was about 2.01 × 10−4 l/mol × vS (R2 = 0.98).
3.4. Influence of the Electrical Energy
3.4.1. In KCl and K2SO4 media
The display of the eliminated percentage at 480 nm taking into account the consumption of electrical energy, shows clearly that the environment chlorinates is more effective than the sulphate one (Fig. 6). For example, for an electrical energy equal to 1.4 KWh/m3, the eliminated percentage is 24% in K2SO4 and 97% in KCl. For the same conditions, the total elimination of OG at 480 nm in the presence of chloride ions is instantaneous. Whereas in the presence of K2SO4, the eliminated percentage increases with time to reach 60% after 4 h of reaction. The electrical energy consumption increases with time. Fig. 6 shows clearly that the experiment consumes more electrical energy in absence of chloride. Indeed, all the dye is eliminated in the KCl medium for a consumption of 0.5 kWh/m3 against 10 kWh/m3 for reaching 60% in free chlorine environment.
3.4.2. Influence of OG concentration
Since the concentration of dye in the industrial waste varies from one period to another, it is necessary to study the variation in the efficiency of electrochemical oxidation treatment depending on the concentration of dye.
During electrochemical treatment of wastewater containing organic pollutants, a parallel evolution reaction of oxygen still occurs causing a decrease in the efficiency of electrochemical oxidation of the considered pollutant. In Table 3, are listed the Orange G degradation data at different concentrations using the same operating conditions. When OG concentration increases from 50 to 75 mg/L, the eliminated percentage increases from 60 to 68.35%. Then, it decreases for a higher OG concentration (100 mg/L) to reach 65% in K2SO4. In mid -rich chlorides, the eliminated percentage of OG does not seem to be affected by the concentrations increase. In addition, the percentage of removed COD increases to rich 90% for 100 mg/L. These results confirm the hypothesis given by several researchers who assert that the electrochemical oxidation process is very favorable for concentrated pollutants discharges [24].
The GCE efficiency is closely related to the COD. By increasing the concentration of the dye, the GCE also increases in both media. So, the treatment of concentrated dye discharges leads to a large mineralization of treated products. This can be explained by the fact that a high concentration of dye in solution decreases the limitation caused by the mass transfer of dye to the surface of the electrode and facilitates its oxidation.
The percentage of removed OG increases with the increase of electric power consumption (Fig. 6). For a concentration of 50 mg/L, a consumption of 0.5 KWh/m3 is sufficient to eliminate all OG from the solution. To achieve the same percentage, it took 9 kWh/m3 for a concentration of 75 mg/L. This amount of electricity consumed causes the removal of 90% of dye at a concentration of 100 mg/L.
The percentage of removed COD increases with increasing OG concentration (Fig. S4). At the beginning of the reaction, the dye concentration does not affect much the percentage of removed COD. By increasing the amount of electricity to 2.7 kWh/m3, the percentage of removed COD increases from 44 to 80% for concentrations of 50 and 100 mg/L, respectively. A quantity of 10.7 kWh/m3 electricity gives 80% of removed COD for all the concentrations tested in this work. This proves that the electrochemical degradation of Orange G on a platinum electrode is very effective even for highly loaded dyes discharge.
In Fig. 7, are presented the GCE variations with the consumption of electrical energy for the three dye concentrations in KCl. The curves are identical in shape; the electric power efficiency decreases with increase in electric power consumption. In chloride middle, the process is effective even for high OG concentrations. The efficiency with electric current is the same for both media with or without chlorides for concentrations of 50 and 75 mg/L. When the dye concentration reached 100 mg/L, this efficiency reached 5.61 in the chloride medium against 3.1 in K2SO4 medium. This is related to the percentage of removed COD, which is important in chloride medium.
4. Conclusions
The main conclusions of this work can be summarized in the following points:
Platinium-anodic oxidation can be successfully used to remove completely all the colour and COD, considering the specific operating conditions (current density, pH and OG concentration).
The comparison of the UV - visible spectra of OG before and after electrochemical oxidation shows the decrease in the intensities of the absorption peaks at 245, 330 and 475 nm with current density. The best results are obtained for 50 mA/Cm2 of current density.
The degradation of Orange G on a platinum electrode is feasible in a wide pH range, so it would be useless to adjust it for a possible application of the method in the industry.
The UV-visible spectra of OG as a function of degradation time suggest that the degradation mechanisms in K2SO4 and KCl medium are not the same.
The presence of chloride ions in solution greatly improves the rate degradation of Orange G. The comparison between kinetic constants demonstrates this behavior.
The kinetics degradation of Orange G at different concentration (50, 75 and 100 mg/L) in the presence and the absence of chloride ions seems to follow the pseudo-first order except for the concentration of 100 mg/L in KCl which follow pseudo second order.
The role of indirect oxidation exceeds 55 times that of the direct oxidation for a 50 mg/L of OG concentration.
The experiment consumes more electrical energy in absence of chloride. Indeed all of the dye is eliminated in the KCl medium for a consumption of 0.5 kWh/m3 against 10 kWh/m3 for reaching 60% in free chlorine environment.
In mid -rich chlorides, the eliminated percentage of OG does not seem to be affected by the concentration’s increase. In addition, the percentage of removed COD increases to rich 90% for 100 mg/L.
By increasing the concentration of the dye, the GCE also increases in both media. So the treatment of concentrated dye discharges leads to a large mineralization of treated products.
Further experiments are in progress in order to demonstrate the applicability of electrochemical technologies for removing dyes contamination from real effluents using electrodes synthesized in laboratory.
The Algerian CNEPRU PROGRAM E01920140015 supports this work (identification and elimination of pollutant by electrochemical process).
References
1. Comninellis C, Chen G. Electrochemistry for the Environment. New York: Springer; 2010.
2. Rao ANS, Venkatarangaiah VT. Metal oxide-coated anodes in wastewater treatment. Environ Sci Pollut Res. 2014;21:3197–3217.
3. Cañizares P, Paz R, Sáez C, Rodrigo MA. Electrochemical oxidation of polyhydroxybenzenes on boron-doped diamond anodes. J Environ Manage. 2009;90:410–420.
4. Martínez-Huitle CA, Ferro S. Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes. Chem Soc Rev. 2006;35:1324–1340.
5. Sires I, Brillas E, Oturan MA, Rodrigo MA, Panizza M. Electrochemical advanced oxidation processes Today and tomorrow. A review. Environ Sci Pollut Res. 2014;21:8336–8367.
6. Brillas E, Sires I. Electrochemical removal of pharmaceuticals from water streams: Reactivity elucidation by mass spectrometry. Trends Anal Chem. 2015;70:112–121.
7. Anglada Á, Urtiaga A, Ortiz I. Contributions of electrochemical oxidation to waste-water treatment: fundamentals and review of applications. J Chem Technol Biotechnol. 2009;84:1747–1755.
8. Martínez-Huitle CA, Andrade LS. Electrocatalysis in wastewater treatment: Recent mechanism advances. Quim Nova. 2011;34:850–858.
9. Martinez-Huitle CA, Rodrigo MA, Sires I, Scialdone OA. Single and coupled electrochemical processes and reactors for the abatement of organic water pollutants: Critical review. Chem Rev. 2015;115:13362–13407.
10. Martínez-Huitle CA, Brillas E. Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review. Appl Catal B. 2009;87:105–145.
11. Panizza M. Importance of electrode material in the electrochemical treatment of wastewater containing organic pollutants. Comninellis C, Chen G, editorsElectrochemistry for the Environment. Berlin: Springer science; 2010. p. 25–54.
12. Liu L, Zhao G, Wu M, Lei Y, Geng R. Electrochemical degradation of chlorobenzene on boron-doped diamond and platinum electrodes. J Hazard Mater. 2009;168:179–186.
13. Tabarra MA, Mallah HA, El Jamal MM. Anodic oxidation of anionic Xanthene dyes at Pt and Bdd electrodes. J Chem Technol Metal. 2014;49:247–253.
14. Chatzisymeon E, Xekoukoulotakis NP, Coz A, Kalogerakis N, Mantzavino DJ. Electrochemical treatment of textile dyes and dyehouse effluents. Hazard Mater. 2006;137:998–1007.
15. Xu XR, Li XZ. Degradation of azo dye orange G in aqueous solutions by persulfate with ferrous ion. Sep Purif Technol. 2010;72:105–111.
16. Madhaven J, Grieser F, Ashokkumar M. Degradation of orange-G by advanced oxidation. Ultrason Sonochem. 2010;17:338–343.
17. Chenini H, Djebbar K, Zendaoui SM, Sehili T, Zouchoune B. Removal of an Azo Dye (Orange G) by various Methods in homogenious phase. Comparative study. Jordan J Chem. 2011;6:307–319.
18. Khenifi A, Bouberka Z, Hamani H, Illikti H, Kameche M, Derriche Z. Decoloration of orange G (OG) using electrochemical reduction. Environ Technol. 2012;33:1081–1088.
19. Yoshida Y, Ogata S, Nakamatsu S, et al. Decoloration of azo dye using atomic hydrogen permeating through a Pt-modified palladized Pd sheet electrode. Electrochim Acta. 1999;45:409–414.
20. Fernandes A, Morao A, Magrinho M, Lopes A, Goncalves I. Electrochemical degradation of C.I. Acid Orange 7. Dyes Pigm. 2004;61:287–296.
21. Wu M, Zhao G, Li M, Liu L, Li D. Graphene oxide wrapped melamine sponge as an efficient and recoverable adsorbent for Pb(II) removal from fly ash leachate. J Hazard Mater. 2009;163:26–34.
22. Panizza M, Michaud PA, Cerisola G, Comninellis Ch. Anodic oxidation of 2-naphthol at boron-doped diamond electrodes. J Electroanal Chem. 2001;507:206–214.
23. Chen X, Chen G. Anodic oxidation of Orange II on Ti/BDD electrode: Variable effects. Sep Purif Technol. 2006;48:45–49.
24. Zhou M, Sarkka H, Sillanpaa M. A comparative experimental study on methyl orange degradation by electrochemical oxidation on BDD and MMO electrodes. Sep Purif Technol. 2011;78:290–297.
25. Panizza M, Zolezzi M, Nicolella C. Biological and electrochemical oxidation of Naphtalenesulfonates. J Chem Technol Biotechnol. 2006;81:225–232.
27. Faouzi M, CaNizares P, Gadri A, et al. Advanced oxidation processes for the treatment of wastes polluted with azoic dyes. Electrochim Acta. 2006;52:325–331.
28. Molina J, Fernandez J, del Rio AI, Bonastre J, Cases F. Characterization of azo dyes on Pt and Pt/polyaniline/dispersed Pt electrodes. Appl Surf Sci. 2011;258:6246–6256.
29. Sales Solano AM, Rocha JHB, Fernandes NS, Da Silva DR, Martinez-Huitle CA. Direct and indirect electrochemical oxidation process for decolourisation treatment of synthetic wastewaters containing dye. Oxid Commun. 2011;34:218–229.
30. Pepio M, Gutierrez-Bouzan MC. Empirical models for the decoloration of dyes in an electrochemical batch cell. Ind Eng Chem Res. 2011;50:8965–8972.
31. Ma XJ, Zhou MH. A comparative study of azo dye decolorization by electro-Fenton in two common electrolytes. J Chem Technol Biotechnol. 2009;84:1544–1549.
32. Rodriguez FA, Mateo MN, Dominguez R, Riveri EP, Gonzalez I. Electrochemical treatment of indigo carmine solutions via active chlorine in a FM01-LC reactor using DSA (Ti/IrO2/SnO2/Sb2O5) Electrodes. ECS Trans. 2011;36:529–538.
33. Gomes L, Miwa DW, Malpass GRP, Motheo AJ. Electrochemical degradation of the dye reactive orange 16 using electrochemical flow-cell. Braz J Chem Soc. 2011;22:1299–1306.
34. Rajkumar K, Muthukumar M. Optimization of electro-oxidation process for the treatment of Reactive Orange 107 using response surface methodology. Environ Sci Pollut Res. 2012;19:148–160.
35. Rajkumar D, Kim JG. Oxidation of various reactive dyes with in situ electro-generated active chlorine for textile dyeing industry wastewater treatment. J Hazard Mater. 2006;136:203–212.
Fig. 1
Schematic diagram of the electrochemical reactor. 1: Radiometer analytical PGZ 301; 2: one-compartment electrochemical cell; 3: platinium anode 4: saturated Ag/AgCl reference electrode; 5: platinium cathode; 6: magnetic stirrer; 7: OG solution.
Fig. 2
Influence of current density on the UV-Visible spectra of OG (50mg/L) after 4 h of treatment in (a) K2SO4 and (b) KCl.
Fig. 3
Percentage of COD and OG removed at 245, 330 and 480 nm function of current density after 4 h of treatment in (a) K2SO4 and (b) KCl.
Fig. 4
Percentage of OG removed as function of time in (a) K2SO4 and (b) KCl.
Fig. 5
Percentage of COD eliminated function of time in K2SO4 and KCl.
Fig. 6
Percentage of OG eliminated at 480 nm as function of the electric power consumption in K2SO4 and KCl.
Fig. 7
Variation of GCE with electrical energy at different concentration of OG in KCl and K2SO4.
Table 1
Physico-chemical Properties of Orange G
Usual and commercial name
Orange G
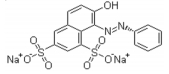
Chemical structure
Molar mass
452,38 g. mol−1
Chemical formula
C16H10N2Na2O7S2
Solubility in water
5 g/100 mL (20°C)
character
Acide
Melting point (°C)
141
λm ax (nm )
475
Use
Textile and dye in bacteriology
Table 2
Kinetic Modeling Results of Orange G Degradation in K2SO4 and KCl Media
[OG] (mg/L)
kobv (S−1)
f
R2
K2SO4
50
6 × 10-5
-
0.98
75
7 × 10-5
-
0.95
100
7 × 10-5
-
0.97
KCl
50
3.4 × 10-3
55.67
0.98
75
6 × 10-4
8.57
0.98
100
-
-
0.57
Table 3
Electrochemical Degradation Parameters of Orange G at Different Concentrations in K2SO4 and KCl Media