1. Introduction
Advanced oxidation processes (AOPs) are widely used in waste-water treatment for the removal of organic and inorganic contaminants or to improve biodegradability of industrial wastewater. Heterogeneous photocatalytic process using TiO2 as catalyst is one of the most promising AOPs. The radicals generated (OH•, O2− •, HOO•) can non-selectively mineralize a large range of organic pollutants which are difficult to eliminate [1]: pharmaceutical compounds [2, 3], dyes [4], pesticides [5], phenols [6], etc. Titanium dioxide (TiO2) photocatalyst was broadly used because of its photo-chemical stability and comparatively low cost [7, 8, 9, 10]. However, the main drawback of the practical use of the TiO2 is the difficulty to separate the TiO2 powder from water. The need of an uneasy and costly final filtration limits the industrial-scale development of these photocatalytic processes. Therefore, to solve this problem, techniques have been developed for immobilizing TiO2 catalyst onto the surface of a solid: sol-gel method [11], hydrothermal method [12], impregnation [13], atomic layer deposition [14], dip coating [15], hydrolysis [16], metal organic chemical vapour deposition [17, 18, 19], carbonization method [20], microwave irradiation [21]. The supporting materials generally suggested for degradation of organic compounds in water are materials such as silica, alumina, zeolites or clays [22], glass, quartz or steel [23, 24], activated carbon [17, 25] or membrane [26]. Improvement of photo-efficiency has not been systematically observed. Among these particle supports, activated carbon (AC) is very promising. Activated carbon adsorbs the pollutants and then releases them onto the surface of TiO2. Consequently, the pollutants are more concentrated around the TiO2 than in the bulk solution leading to an increase in the degradation rate of the pollutants [27, 28]. The intermediates produced during degradation could also be adsorbed onto activated carbon and then be oxidized. Other authors [29, 30, 31] have reported a synergistic effect for simple mixtures of AC and TiO2, pollutants being more rapidly photodegraded in the mixed system which contains activated carbon. This so-called synergetic effect has been explained by the formation of a common contact interface between the different solid phases, in which AC acts as an efficient adsorption trap to the organic compounds. The pollutant is then more efficiently transferred to the TiO2 surface [32, 33, 34].
The main fundamental problem in using TiO2 as photocatalyst is the energy loss in the electron-hole recombination which results in a lower efficiency of the degradation. Hence the prevention of electron-hole recombination becomes very important, the addition of electron acceptors into the reaction media is recommended: molecular oxygen present in air [35, 36] or H2O2 have been employed as effective electron acceptor in most photocatalysis applications. H2O2 could serve as electron scavenger to prevent the recombination and, in addition, generate more hydroxyl radicals [2]. In most of cases, the presence of H2O2 improved the photocatalytic oxidation performances [2, 27, 37] some authors reported a negative effect of H2O2 [38, 39, 40, 41]. Ilisz et al. [42] mainly observed the formation of primary oxidation products (hydroquinone, catechol and presumed aliphatics) at low concentration of H2O2 (< 0.01 mol/L) while hydroquinone and catechol concentration strongly decreased and ring-opening products were predominant at high H2O2 concentration (> 0.05 mol/L), more interesting in a complete degradation objective. Adan et al. [43] proposed the optimal molar ratio between concentrations of hydrogen peroxide and pollutant to be between 10 and 100.
In this work, phenol has been chosen as a model molecule representing hazardous phenol compounds in the industry. Indeed, phenolic compounds are identified as highly toxic compounds and non-biodegradable molecules. To degrade these compounds, a photocatalytic process has been implemented using commercial composite multilayer material TiO2/AC. The photocatalytic activity of this material was evaluated in two different reactors and two different reaction modes. First, a sequential method of water treatment was carried out involving adsorption then photocatalytic oxidation in a loop reactor, with or without H2O2 to examine the improvement of photocatalysis by H2O2 addition. In this configuration, the water treatment was not achieved by oxidation but by adsorption, the oxidative step being needed only for pollutant degradation and in situ activated carbon regeneration. Then, the performance of the composite material was studied in a continuous annular reactor with simultaneous adsorption and photocatalysis.
2. Materials and Methods
2.1. Materials
Phenol (Bioxtra 99.5%) and hydrogene peroxide (30% w/w) were supplied by Sigma Aldrich.
Ahlstrom directly supplied the composite material TiO2/AC, and separately the TiO2 catalyst (PC 500, Millenium, anatase > 99%) and the AC. The composite material was a tissue composed with a mixture of synthetic fibers, acrylic polymer and granular activated carbon, a mixture of TiO2 is coated on one side (EP1069950B1 European patent). The weight composition of this tissue is 4.5% TiO2, 63.5% AC, 27.5% fibers, 4.5% SiO2. SiO2 was used as an inorganic binder for titanium deposited on the paper fibers. It is transparent with UV light and photo-stable. The granular activated carbon made from coconut with particle diameter in the range 250–600 μm, and its specific surface was 1,065 m2/g. This activated carbon was mainly microporous (microporous and mesoporous volumes of 0.46 and 0.046 cm3/g respectively). The diameter of the TiO2 crystals was between 5 and 10 nm and the specific surface area SBET about 320 m2/g.
2.2. Analytical Methods
2.2.1. Solid analysis
The textural characterization was deduced from nitrogen adsorption at 77 K (ASAP 2010M; Micromeritics, Norcross, GA, USA). The specific surface area SBET was determined from the BET method for relative pressure range (p/p0) of 0.01–0.20 [44]. Methods from Horvath and Kawazoe [43] and Barrett et al. [44], referred as HK and BJH, were employed to assess the micropore and mesopore volumes respectively.
2.2.2. Liquid analysis
Phenol concentration measurements were performed by a high performance liquid chromatography equipment with UV detection at 254 nm (UV2000 dual wavelength; Thermo Finnigan, Les Ullis, France) using a C18 column (Prontosil, 4 mm× 250 mm i.d., 5 μm particles, I.C.S., Lapeyrouse-Fossat, France), with a mobile phase composed of acidified deionized water (W) (water acidified by H3PO4; pH = 2.2) and Acetonitrile (A) fed at 1 mL/min. The detector wavelength was set to 254 nm and the temperature of the column maintained at 30°C. For the adsorption step, an isocratic method was utilised (volumic composition Aqueous/Organic is 60/40). For the oxidation step, the separation of phenol from the oxidation intermediates was achieved with a mobile phase of variable volume composition W/A programmed at 1 mL/min (0–3 min: W only; 3–16 min: gradient W/A from 100/0 to 60/40; 16–25 min: 60/40).
The chemical oxygen demand (COD) was measured using colorimetric or digestion method with Hach equipment: a digital reactor block (DRB 200 HACH LANGE; Thermo Fisher Scientific, Illkirch, France), a spectrophotometer (DR/2500 HACH LANGE; Thermo Fisher Scientific, Illkirch, France) using 0–1,500 mgO2/L range Hach tubes (potassium dichromate, salt of silver and mercury, sulfuric acid). The precision of the method was assessed with standard solutions and showed a standard deviation of less than 5%.
The total organic carbon (TOC) was also measured during photo-catalytic oxidation step. First, the inorganic carbon present in the solution was eliminated with concentrated phosphoric acid (84%) and the solution was degassed by a current of nitrogen. The sample was then injected in a TOC-meter (TC Multi Analyser 2100 N/C; Analytic Jena, France) where the organic molecules were totally oxidized at 850°C, over a platinum catalyst. The quantity of CO2 released by the reaction was then measured by infrared spectrometry (IR). Total organic carbon analysis was made when H2O2 was added in the oxidation step. Indeed, COD is no longer possible due to H2O2 interference in the dichromate method.
2.3. Experimental Set-ups and Procedures
2.3.1. Batch adsorption
Isotherms of the original granular AC and TiO2/AC material were performed. In brown flasks, 0.5 g of adsorbent (AC or TiO2/AC) was added to 100 mL of phenol solutions (0.5–5.0 g/L concentration range). The suspensions were left under stirring in a thermo-regulated bath at 25°C for 8 days to ensure equilibrium. The solutions were not buffered, the final pH was in the range of 4 to 5. Then, the solutions were filtered on 0.25 μm nylon filter membranes before analysis [25]. The amount of adsorbed phenol was deduced from initial C0 and final HPLC measurements of concentrations in the liquid phase.
2.3.2. The loop reactor
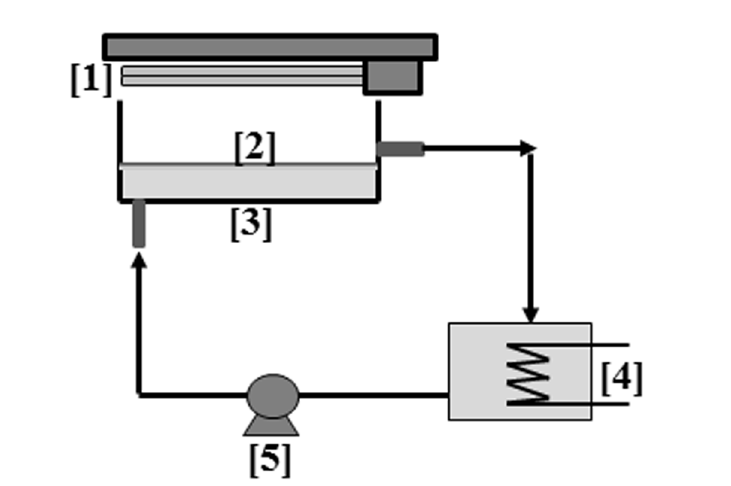
The 2 L batch loop reactor (Fig. 1) composed of a reactor section (0.65 L) and a recycling tank (1.35 L) was designed to operate with a sheet of composite TiO2/AC (dimension: 360 mm × 120 mm × 3 mm; weight: 17 g with 10.9 gAC and 0.8 gTiO2). The sheet was maintained barely immersed at the top of the flowing aqueous solution of phenol (C0 = 0.88 g/L) which was circulated by a peristatic pump (MasterflexL/S; Thermo Fisher Scientific, Illkirch, France) at a constant flow rate (1,000 mL/min). The recycling tank was cooled by thermostatic water bath at 25°C (Julabo F12-ED; SELI, Toulouse, France). The equipment did not include the air supply as enough gas-liquid surface was available under both the composite sheet and the recycling tank to keep dissolved oxygen in the liquid. The range of emission of the 2 lamps (PL-L 24W; Philips, Bossee, France) was 340–400 nm with a maximum at 365 nm. The mean value of the light intensity measured with a radiometer (model RS232 Lutron; Thermo Fisher Scientific, Illkirch, France) was 55 W/m2 at the surface of the composite material.
During the sequential process, two successive steps were achieved: adsorption then photo-oxidation corresponding to one cycle. The adsorption step was carried out in the dark during 4 days to approach the maximum loading corresponding to the adsorption equilibrium. Then, the oxidation step was started by switching on the UV lamps for 3 days. During the two steps samples were taken at regular time intervals and analysed by HPLC for determining the phenol concentration. A part of the samples was also used for measuring COD or TOC. TOC measurements have been chosen in presence of H2O2 in the oxidation step. After each oxidation step, the reactor was emptied from the oxidized solution of phenol and then filled by 2 L of a new one at same initial concentration (C0 = 0.88 g/L) for starting a next cycle. High initial concentration of phenol has been chosen to quickly saturate the composite material and so show its regeneration.
2.2.3. Annular continuous reactor
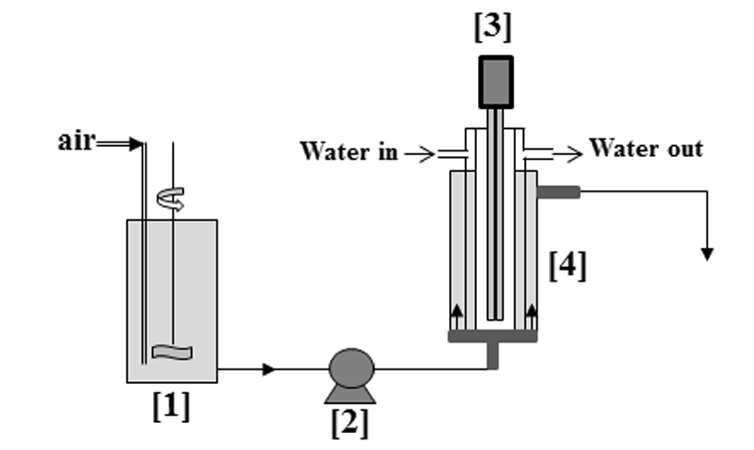
Experiments were carried out in an annular reactor of 120 mL shown in Fig. 2. On the central axis, the UV lamp was placed in a jacketed thermo-regulated cylinder (25°C). The mean value of the light intensity measured with the radiometer was 72 W/m (at the inside surface of the jacket cylinder). A peristaltic pump with a flow rate of 2 mL/min was used circulate the solution from the inlet tank to the annular reactor. The size of the sheet of composite TiO2/AC was chosen to entirely fill the annular space (mTiO2/AC = 12.7 g, mTiO2 = 0.6 g). The experimental protocol was as follows. The composite material TiO2/AC was pre-loaded in a beaker for 3 days using a solution of phenol (C0 = 0.209 g/L) until the equilibrium was reached (Cfinal = 0.018 g/L). After this dark adsorption period, the oxidation process was then started using this loaded composite material as a pseudo-fixed bed in the annular reactor. As soon as the lamp was switched on, this reactor was continuously fed with a phenol solution, Cinlet = 0.018 g/L. The samples were taken each hour at the exit of the reactor via a sampling port. The experiment was conducted at 25°C. All samples were analysed by HPLC to determine the phenol concentrations.
3. Results and Discussion
3.1. Adsorption Isotherms
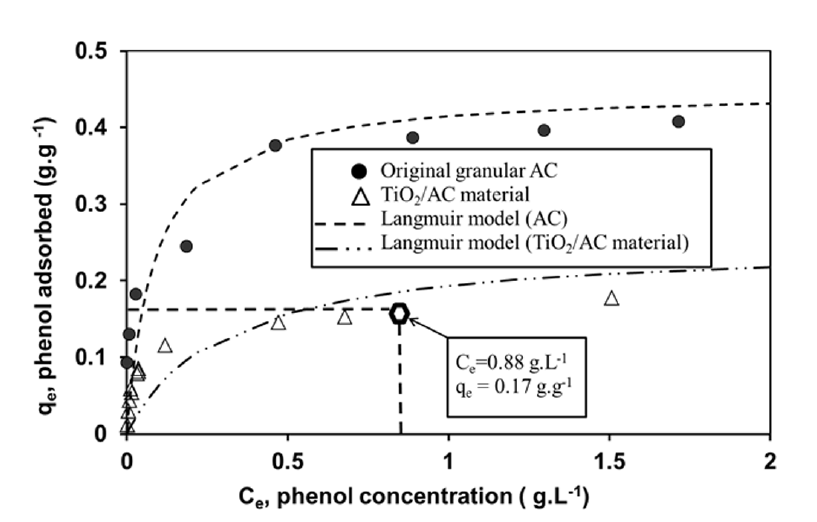
The adsorption isotherms of the original granular AC and TiO2/AC material are presented in Fig. 3. Both isotherms exhibited the same shape, corresponding to type I of international union of pure applied chemistry classification (1985) as expected regarding the high AC microporosity (monolayer adsorption). Calculation of isotherm parameters has been performed using linearization of the Langmuir model equation. The Langmuir parameters given in Table 1 showed a higher value of the maximum adsorption capacity qmax (0.45 g/g) for the activated carbon alone. This saturation capacity was more important than the reported values in the literature for phenol adsorption. Usual values of qmax are between 0.20 and 0.35 g/gAC under ambient temperature and unbuffered conditions showing the excellent performances of this activated carbon [47, 48, 49, 50, 51].
The composite material adsorption capacity is of course lower -as AC corresponds to only 63.5% of this material weight- but still very convenient. Note that in addition, when incorporated in the tissue, AC seems to lose about 12% of its specific adsorption capacity which could be due to partial pore clogging.
3.2. Sequential Process in Loop Reactor
3.2.1. Sequential process – 5 cycles without H2O2
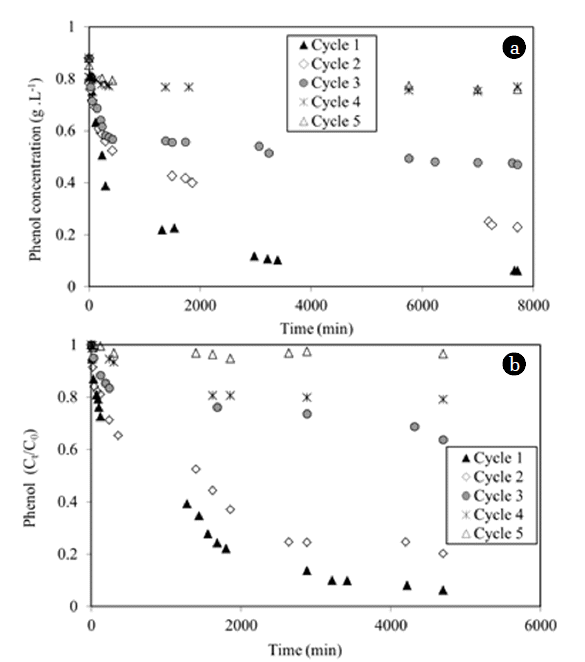
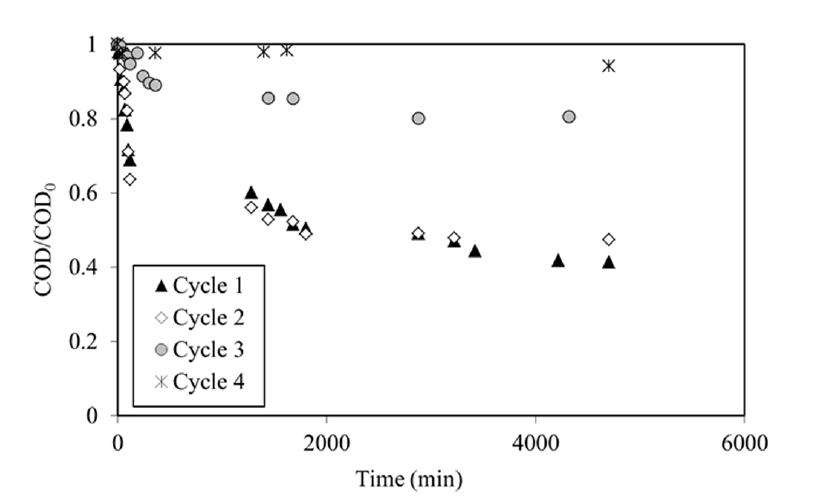
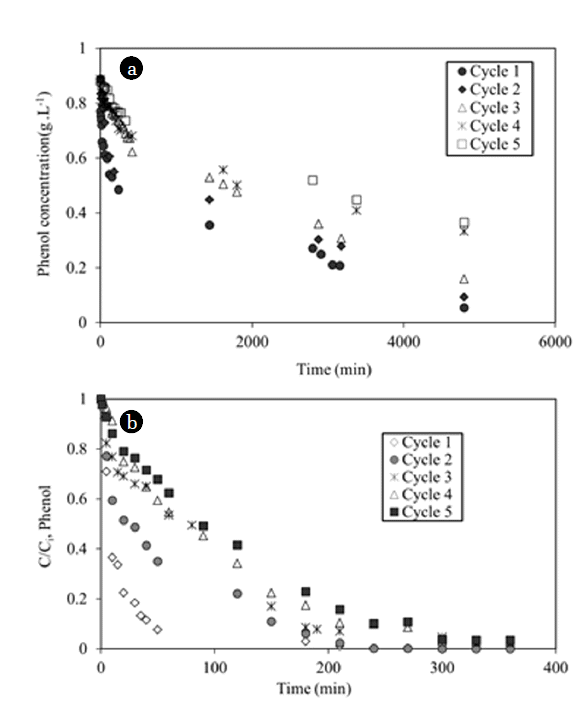
Preliminary experiment with phenol and UV light was performed. Direct photolysis of phenol during 3,500 min did not change the concentration as expected from the absorbance spectrum of phenol (no absorbance above 310 nm). Five cycles of adsorption-photo-catalytic regeneration have been carried out with the composite material (Fig. 4(a) and (b)). In comparison with classic adsorption kinetics with granular carbon, the kinetics were very slow probably due to the diffusion limitation of the phenol inside the layer of natural and synthetic fibers containing AC. Table 2 showed a gradually decrease of the quantities adsorbed and the regeneration efficiency Re calculated from Eq. (1).
The total quantity of adsorbed phenol (0.218 g/g) was 40% higher than the theoretical maximum adsorbed amount for a concentration equal to 0.88 g/L (0.17 g/g, see isotherm on Fig. 3) proving the effective regeneration of the composite material. The final degraded fraction decreased from 95% at cycle 1 to 1% at cycle 5 due to an incomplete regeneration of the material. The first-order kinetic model (Eq. (2)) was used to describe the kinetics of phenol degradation (Fig. 4(b)) and to obtain the values of the apparent constant of degradation at each cycle.
The model fitted well until about one hour, then the presence of an important quantity of intermediates (hydroquinone, catechol, benzoquinone, aliphatic compounds, etc.) and the competitive adsorption between phenol and these intermediates might change kinetics [52]. Consequently, the calculation of kapp was done using the first part of these kinetics. Decrease of the successive apparent contants kapp confirmed the continuous degradation of the regeneration efficiency and the pseudo-first order of the kinetic (depending on total concentration). This phenomenon was classically observed in photocatalysis when high quantity of organic molecules had to be degraded [53]. Above a certain level of phenol concentration, the catalyst surface becomes saturated, and this may even lead to a decrease in the observed rates [54]. The COD evolution was represented in Fig. 5. The degradation did not exceed 5% for the 4th cycle. For the first cycle, even if 95% of the phenol was removed in the liquid phase, 40% of the initial COD remained in solution due to the reaction intermediates. At the end of each oxidation step, the contribution of phenol to the COD was calculated using phenol concentration (HPLC): 15%, 42%, 80% and 84% for the 1st, 2nd, 3rd and 4th cycles respectively (Table 2). These results confirmed that the conditions of oxidation were degraded from the second cycle.
At the end of the 5th cycle, the activated carbon inside the composite material was analyzed. The surface area SBET was reduced by 80% (from 1,065 m2/g to 208 m2/g) and the microporous volume by 84% (from 0.460 cm3/g to 0.076 cm3/g). An important quantity of phenol and oxidation intermediates were accumulated in the micropores without being further oxidized. These molecules deeply adsorbed in these pores did not migrate from the activated carbon to the catalyst to be degraded. The expected mechanism (adsorption of phenol → phenol diffusion from AC to TiO2 → total phenol mineralization under UV → total regeneration of the composite material) was no longer taking place. At the photo-catalyst surface, the phenol and intermediates remained concentrated and cycle after cycle more organic compounds get adsorbed. The photocatalytic activity decreased due to the accumulation of reaction intermediates formed at the surface as a result of partial oxidation of phenol. The presence of these molecules reduced number of reaction sites and reactivity with time. The catalyst used presented a low selectivity toward total oxidation with respect to partial oxidations [14]. This loss of activity might suggest a poisoning of the surface of the catalyst. Another explanation could be proposed: at the surface of the catalyst, these intermediates were probably in competition with H2O and O2 on the adsorption sites. Then less molecules of water and oxygen would be adsorbed at the surface of the catalyst, therefore, an increase of the recombination of the electron/hole pairs could occur. This phenomenon could explain a lower production of oxidative species (OH• and O2− •) and an additional loss of photo-activity.
3.2.2. Sequential process – 5 cycles with H2O2
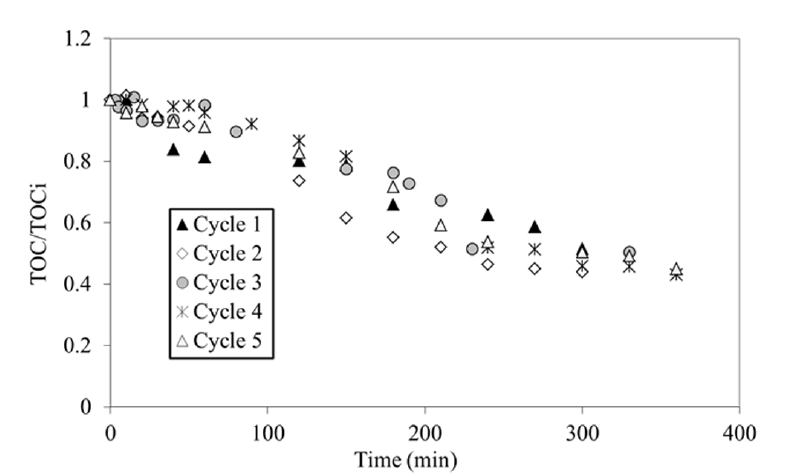
In order to improve the oxidation process the effect of H2O2 addition on the rate of phenol degradation was investigated. At the beginning of each oxidation step, 50 mL of H2O2 (30% weight) were added to the solution. For the first oxidation, the corresponding initial H2O2/phenol molar ratio = 24 was almost twice the stoichiometric amount of hydrogen peroxide for complete mineralization, in agreement with the values proposed by Adan et al. [43] in the range 10 to 100). In presence of H2O2, Fig. 6(a) showed an effective adsorption for all of the five cycles. The amount of adsorbed phenol decreased, but much more slowly than without H2O2 and the regeneration efficiency remained superior to 60%: the regeneration was still partly achieved even after 5 cycles (Table 2). The total quantity of phenol adsorbed during 5 cycles (0.389 g/g) was 140% superior to the theoretical maximum amount adsorbed (see Fig. 3) and almost twice the quantity eliminates without H2O2. From these results, the coupling of H2O2 and TiO2 significantly improved the performance of the AC regeneration. As shown in Fig. 6(b), the phenol degradation was only slightly decreased from 100% to 96%. The pseudo first-order kinetic model (Eq. (2)) fitted well, the apparent constant kapp were between 4 and 12 times higher than without H2O2 (Table 2). The intermediates degradation has gone further as shown by the evolution of the TOC in Fig. 7. Indeed, the TOC conversion rate was between 60% and 70%. The comparison between total TOC and phenol contribution TOC phenol showed an important presence of intermediates (Table 2) showing the partial oxidation of the phenol. This is in agreement with Zhang et al. [55] who reported rather high values of TOC even at total degradation of the aromatic molecules. These intermediates product are thought to be aliphalic acids known to be difficult to oxidize [56].
For the first cycle, the synergetic factor S, calculated with Eq. (3), was equal to 7.5 confirming the better photocatalytic performance with H2O2.
Where:
kapp (with H2O2) is the value of kapp with the composite material, with H2O2 and with UV
kapp (without H2O2) is the value of kapp with the composite material and with UV
kapp (alone) is the value of kapp without the composite material and with UV (reference).
This better performance was due to two additional pathways of •OH production in presence of H2O2 [41, 57]:
The quantity of added H2O2 was chosen in agreement with literature to avoid the negative effect of H2O2. Indeed, introduced in excess, H2O2 could react with hydroxyl radicals and the holes (reactions (7) and (8)): a weaker oxidizing radical •HO2 being produced, inducing a decrease of the photocatalytic performances [2].
After the 5th cycle, the AC present in the composite material was analyzed and compared to the virgin one: the surface area SBET was reduced by 47% (from 1,065 m2/g to 562 m2/g) and the microporous volume by 75% (from 0.460 cm3/g to 0.113 cm3/g). In comparison with experiments without H2O2, this better preservation of surface area and porous volume proved phenol and intermediates to be less accumulated in the pores leading to more efficient regeneration.
3.3. Continuous Annular Reactor
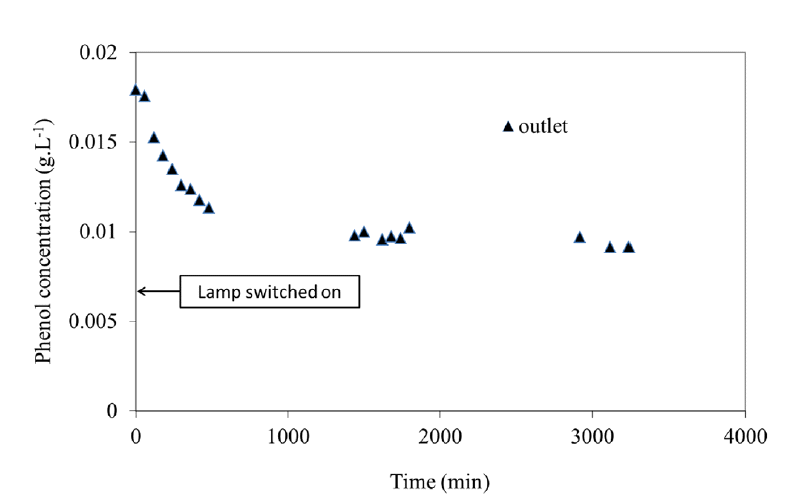
In continuous mode, adsorption and photocatalytic oxidation operated simultaneously. Under UV light, Fig. 8 shows a transient behaviour with initial decreasing phenol concentration due to the added photocatalytic activity. Then the steady state was obtained corresponding to equilibrium between adsorption and oxidation and proving the composite to get a stable behaviour without noticeable deactivation. In these conditions, 55% of the phenol was removed corresponding to an overall reaction rate of 5 10−6 (gphenol/gTiO2) s−1.
4. Conclusions
This study was conducted to put on the map an innovative process to degrade aromatic molecules. To offer an economically interesting process, the conditions were: (1) to use a commercial material and cheap UV lamps; (2) to prolong the adsorption activity of the material with in situ regeneration.
A first implementation was involved using a loop reactor. This sequential process alternated 5 cycles with a step of adsorption in dark and a step of photocatalytic oxidation under UV irradiation. Without H2O2, the regeneration of the composite multilayer material became ineffective from the third cycle on. Cycle after cycle, more pollutants were present at the surface of the composite material and in the solution. The apparent constants decreased drastically, reaction intermediates accumulated on the surface of the photo-catalyst, reducing number of reaction sites and reactivity with time. After several uses, this accumulation induced a poisoning of the catalyst. The presence of H2O2 improved the conditions of regeneration. For the 5 cycles, the phenol degradation was between 100% and 96% and the TOC conversion rate was between 60% and 70%. At industrial scale, the process could be implemented as follow: (1) a step of continuous adsorption by controlling the outlet concentration; (2) a step of regeneration in batch mode using a small volume of water and in presence of UV and H2O2 controlling and optimizing the quantity of UV light, the catalyst loading and the duration of irradiation.